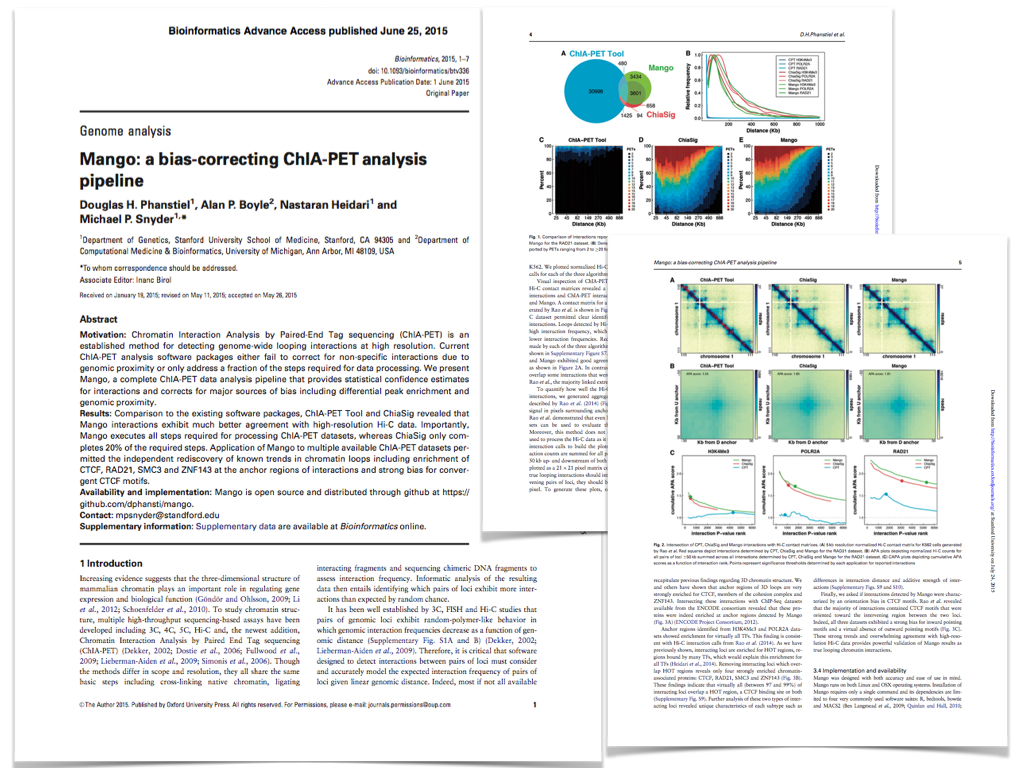
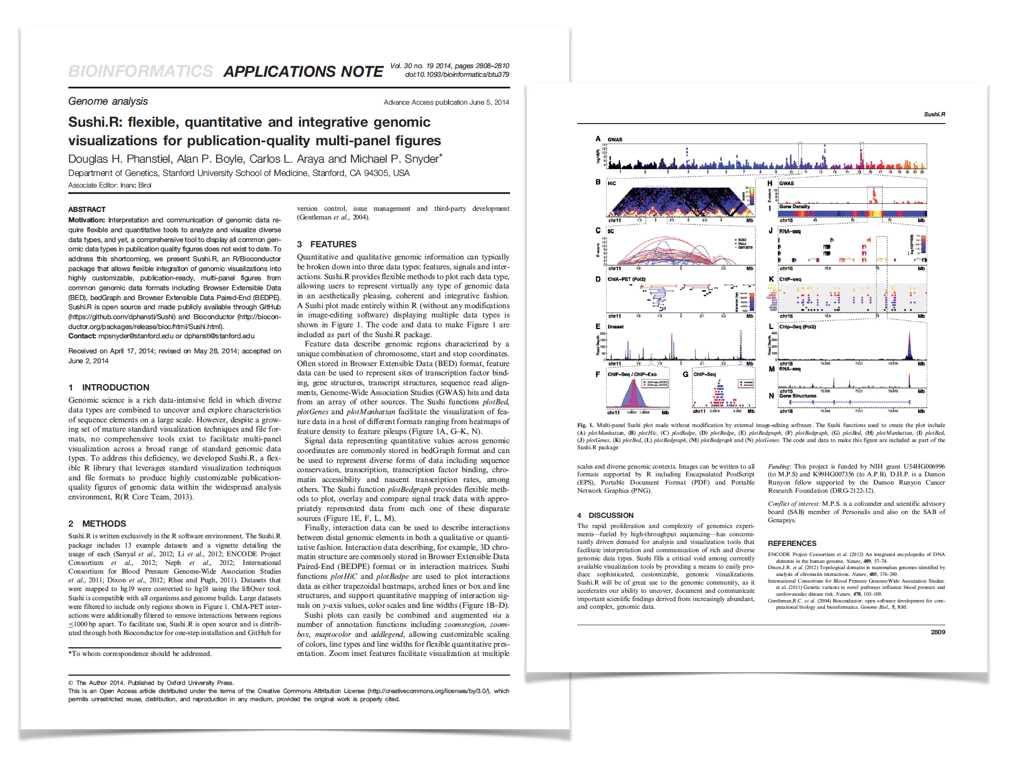
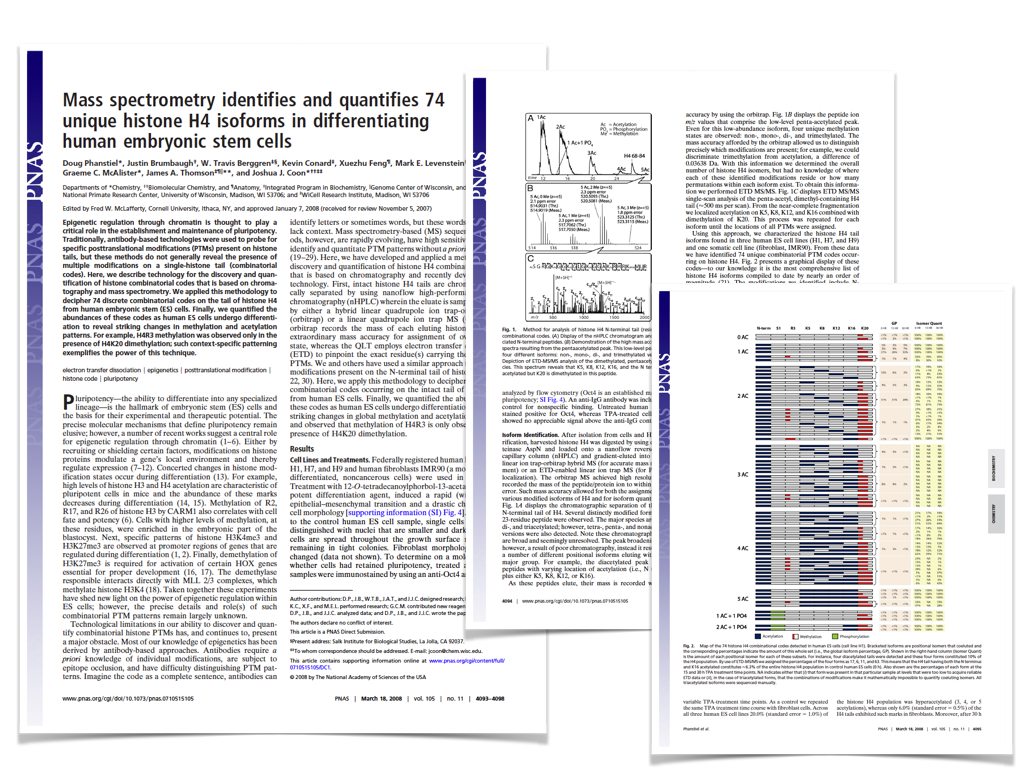
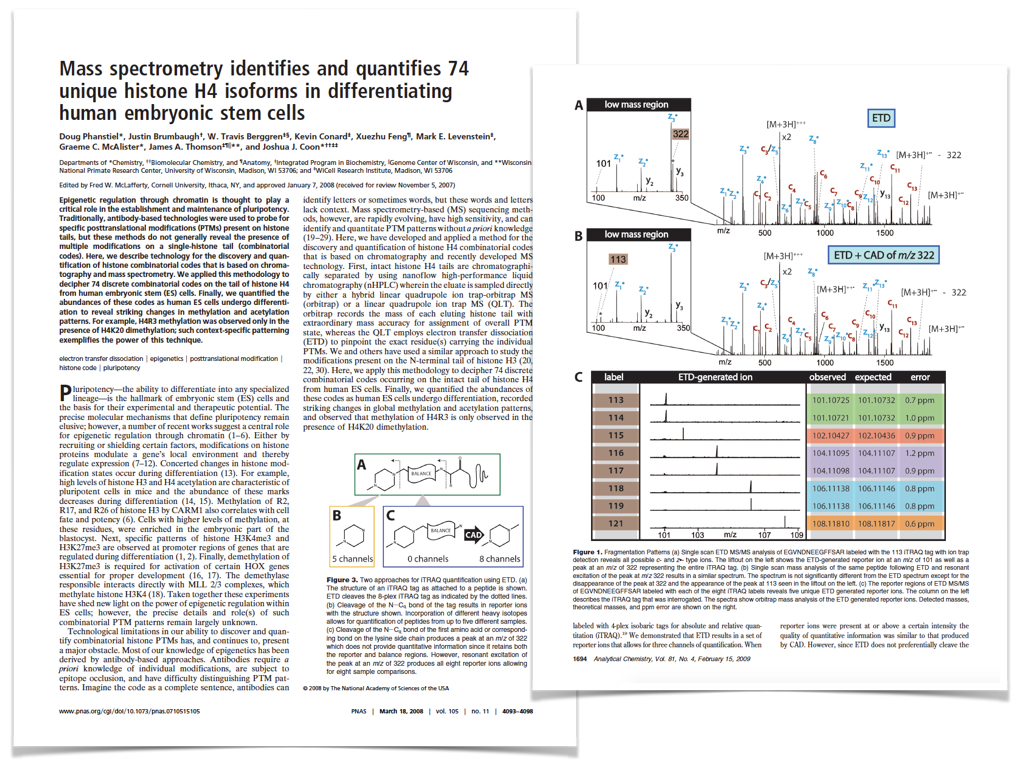
Publications
University of North Carolina at Chapel Hill

Coral: Clear and Customizable Visualization of Human Kinome Data
Metz, K.S., Deoudes, E.M., Berginsky, M.E., Jimenez-Ruiz, I., Aksoy, B.A., Hammerbacher, J., Gomez, S.M., Phanstiel, D.H. Cell Systems, 2018.
Protein kinases represent one of the largest gene families in eukaryotes and play roles in a wide range of cell signaling processes and human diseases. Current tools for visualizing kinase data in the context of the human kinome superfamily are limited to encoding data through the addition of nodes to a low-resolution image of the kinome tree. We present Coral, a user-friendly interactive web application for visualizing both quantitative and qualitative data. Unlike previous tools, Coral can encode data in three features (node color, node size, and branch color), allows three modes of kinome visualization (the traditional kinome tree as well as radial and dynamic force networks), and generates high-resolution scalable vector graphics files suitable for publication without the need for refinement using graphics editing software. Due to its user-friendly, interactive, and highly customizable design, Coral is broadly applicable to high-throughput studies of the human kinome. The source code and web application are available at github.com/dphansti/CORAL and phanstiel-lab.med.unc.edu/Coral, respectively.
paper pdf preprintStanford University
-
Topological organization and dynamic regulation of human tRNA genes during macrophage differentiation.
Van Bortle K, Phanstiel DH, Snyder MP. Genome Biology, 2017.

Static and Dynamic DNA Loops form AP-1-Bound Activation Hubs during Macrophage Development
Phanstiel, D.H.*, Van Bortle, K.*, Spacek D.V., Hess G.T., Saad Shamim M., Machol I., Love M.I., Lieberman Aiden E., Bassik M.C., Snyder, M.P. Molecular Cell, 2017.
The three-dimensional arrangement of the human genome comprises a complex network of structural and regulatory chromatin loops important for coordinating changes in transcription during human development. To better understand the mechanisms underlying context-specific 3D chromatin structure and transcription during cellular differentiation, we generated comprehensive in situ Hi-C maps of DNA loops in human monocytes and differentiated macrophages. We demonstrate that dynamic looping events are regulatory rather than structural in nature and uncover widespread coordination of dynamic enhancer activity at preformed and acquired DNA loops. Enhancer-bound loop formation and enhancer-activation of preformed loops together form multi-loop activation hubs at key macrophage genes. Activation hubs connect 3.4 enhancers per promoter and exhibit a strong enrichment for Activator Protein 1 (AP-1) binding events, suggesting multi-loop activation hubs involving cell-type specific transcription factors may represent an important class of regulatory chromatin structures for the spatiotemporal control of transcription.
paper pdf preprint